APIGENINA - jak odkryłam, że pomaga mojemu dziecku na tiki?
 W 2017 roku byłam już bardzo zmęczona poszukiwaniem pomocy dla mojego chorego synka, gdyż tiki i zaburzenia obsesyjno-kompulsywne dla lekarzy nie stanowiły problemu i miały być rozwiązane przez jedno lekarstwo...rispolept.
W 2017 roku byłam już bardzo zmęczona poszukiwaniem pomocy dla mojego chorego synka, gdyż tiki i zaburzenia obsesyjno-kompulsywne dla lekarzy nie stanowiły problemu i miały być rozwiązane przez jedno lekarstwo...rispolept.
Koprolalia? Proszę oduczyć syna! Moczenie nocne? Błędy wychowawcze! Lęk separacyjny? Nadopiekuńczość! Wymieniać dalej? Odbijając się od ściany, działając w pojedynkę starałam się poznać jaka droga prowadzi od błędnej odpowiedzi układu odpornościowego do powstania tików, obsesji oraz kompulsji.
Naukowcy uważają, że u niektórych osób częste infekcje wywołują nieprawidłową odpowiedź immunologiczną, która powoduje, że przeciwciała atakują zdrowe komórki w mózgu. Ten autoimmunologiczny atak na mózg może spowodować stan zapalny i wystąpienie objawów neuropsychiatrycznych, w tym tików. Tiki to krótkie, powtarzające się, mimowolne ruchy lub dźwięki, takie jak grymasy twarzy, szarpanie ramionami lub chrząkanie, czkawka, a nawet pociąganie nosem, czy marszczenie czoła. A więc zaangażowana jest w to motoryka naszego ciała, funkcje te obsługują w mózgu jądra podstawy.
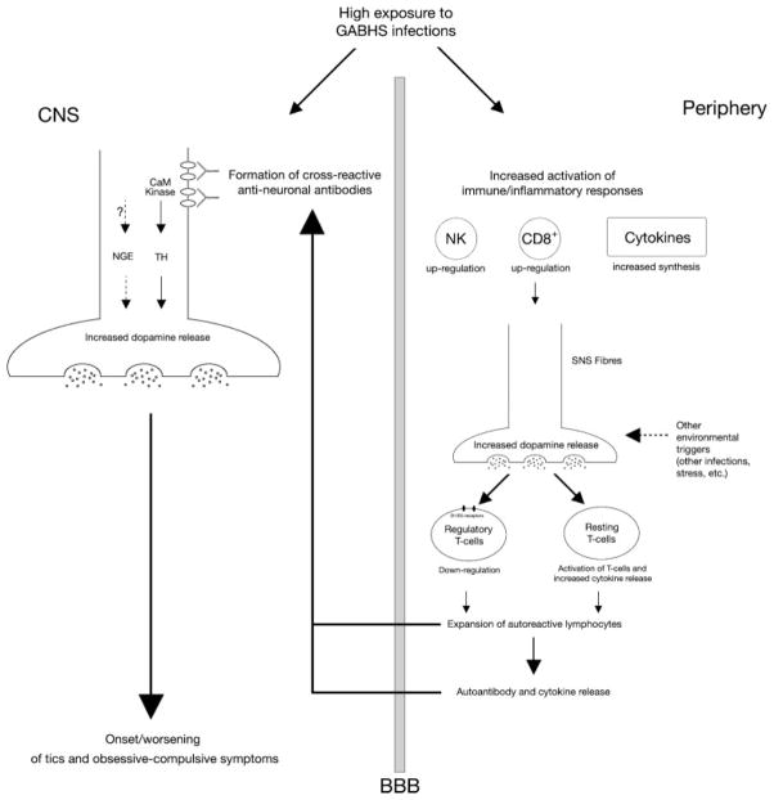
Zespół PANDAS ma wieloczynnikową etiologię, w której czynniki genetyczne, środowiskowe, immunologiczne i hormonalne współdziałają, razem tworząc mieszankę wybuchową. Zwiększona aktywacja odpowiedzi immunologicznych jest spowodowana zmianami w profilach ekspresji genów obwodowych komórek odpornościowych, zmienną częstością występowania subpopulacji limfocytów i syntezą cząsteczek efektorowych układu odpornościowego. Na wzmożoną aktywność mechanizmów komórkowych wskazuje zwiększona ekspresja genów kontrolujących komórki NK i cytotoksyczne limfocyty T, zwiększone stężenie w osoczu niektórych cytokin prozapalnych, które koreluje z ciężkością choroby oraz zwiększoną syntezę przeciwciał przeciwneuronalnych. Wreszcie, na ogólną predyspozycję do odpowiedzi autoimmunologicznych u pacjentów z PANDAS wskazuje zmniejszona częstość występowania limfocytów T regulatorowych, które indukują tolerancję na własne antygeny.
Sugeruje się, że czynniki środowiskowe powodujące aktywację układu odpornościowego odgrywają rolę już we wczesnym dzieciństwie.
Krótko przypomnę kryteria jakie są potrzebne do postawienia diagnozy PANDAS (pediatryczne autoimmunologiczne zaburzenia neuropsychiatryczne związane z zakażenie paciorkowcami).
1. początek przed okresem dojrzewania - pamiętaj, że można go pomylić np. z buntem dwulatka, czy "niewydolnością rodzicielską"...
2. przewlekłe zaburzenie tikowe i/lub zespół obsesji i kompulsji
3. nagłe okresy ciężkich zaostrzeń („flares”) – przebieg nawracająco-remisyjny;
4. czasowy związek początku i jednego lub większej liczby zaostrzeń z klinicznymi objawami zakażenia paciorkowcem;
5. obecność objawów neurologicznych, takich jak zmniejszona koordynacja ruchowa lub zwiększona nadpobudliwość ruchowa
Podczas przeglądu badań epidemiologicznych i opisów przypadków ważne jest, aby pamiętać, że te rygorystyczne kryteria nie zawsze są stosowane. W szczególności kryterium numer pięć może być problematyczne, ponieważ subtelne ruchy pląsawicze są dość powszechne u dzieci w wieku 3–12 lat.
Z drugiej strony u pacjentów z pląsawicą Sydenhama mogą czasami pojawić się tiki. Ponadto rozróżnienie między pląsawicą, a tikami może czasami być niejasne, trudne, co stwarza problem w diagnostyce różnicowej między pląsawicą i PANDAS. Zarówno pląsawica, jak i tiki składają się z szybkich, nierytmicznych, mimowolnych ruchów, ale chociaż ruchy pląsawicze są niestereotypowe, nieregularne w rozmieszczeniu i prawie stale wymykają się kontroli wolicjonalnej, tiki są stereotypowe, często poprzedzone wrażeniami przepowiadającymi i częściowo pod kontrolą wolicjonalną. Jeśli chodzi o kryterium 2, sugeruje się, że wiele innych objawów neurologicznych i psychiatrycznych występuje w PANDAS. Wreszcie kryterium nr 4 jest problematyczne, ponieważ liczne opisy przypadków PANDAS opierają diagnozę na pojedynczym zdarzeniu, a nie na wielokrotnych zaostrzeniach. Warto jednak zauważyć, że pląsawicę można z pewnością rozpoznać na podstawie dramatycznego pojawienia się początkowego epizodu. Natomiast większość badań PANDAS nie skupiała się na początkowym obrazie klinicznym.
Związek między infekcjami paciorkowcowymi, a wystąpieniem lub pogorszeniem zaburzeń obsesyjno-kompulsyjnych został już dobrze udokumentowany. Badania pokazują wyższe miana przeciwciał przeciw paciorkowcom i dodatnią korelację między mianem przeciwciał, a nasileniem objawów klinicznych u dzieci z zaburzeniami obsesyjno-kompulsyjnymi lub tikami. Pamiętajmy, że dzieci z niedoborami odporności stanowią także dużą grupę pacjentów zapadających na zespół PANDAS, a co za tym idzie ich odpowiedź immunologiczna nie jest adekwatnie wyrażona. Infekcja była, a przeciwciał brak.
Obserwacje kliniczne sugerują, że zaburzenia te są wrażliwe na stres psychospołeczny.
Nie od dziś wiemy, że stres moduluje odpowiedź immunologiczną, czyniąc nas bardziej podatnymi na infekcje.
Zakażenie paciorkowcem jest głównym początkowym zdarzeniem wywołującym odpowiedź autoimmunologiczną, przyczyniającym się do wystąpienia zespołu PANDAS. Jednakże udokumentowano, że zaostrzenie tików może być wywołane także przez inne czynniki zakaźne (np. wirus opryszczki pospolitej, wirus ospy wietrznej i półpaśca, ludzki wirus niedoboru odporności, wirusy z rodziny Herpes, Borrelia burgdorferi, Mycoplasma pneumoniae i inne), a także zidentyfikowano szereg innych czynników wywołujących, w tym zapalenie zatok, dziąseł, czy nawet wyrzynające się zęby!
Choroby autoimmunologiczne powstają na skutek załamania procesów tolerancji immunologicznej, które hamują aktywność potencjalnie autoreaktywnych limfocytów T i B, ograniczając w ten sposób odpowiedź immunologiczną skierowaną w stronę „ja”. Jeden z mechanizmów tolerancji obwodowej obejmuje podzbiór limfocytów T, określonych jako limfocyty T regulatorowe (Treg) (lub CD4+CD25+), które są silnymi inhibitorami limfocytów B, CD4+ i limfocytów T CD8+. Wykrywana jest zmniejszona liczba Treg w chorobach autoimmunologicznych, takich jak toczeń rumieniowaty, reumatoidalne zapalenie stawów, cukrzyca typu 1 i stwardnienie rozsiane. Występuje również zwiększona częstość występowania aktywowanych limfocytów B.
Receptory dopaminy mają działanie immunomodulujące, a aktywacja ich zmniejsza aktywność supresyjną oraz zdolności adhezyjne i migracyjne limfocytów Treg.
Cząsteczki efektorowe (cytokiny, chemokiny, cząsteczki adhezyjne) modulują aktywność wrodzonych i nabytych komórek immunokompetentnych. Odpowiedzi adaptacyjne są regulowane przez limfocyty pomocnicze T typu 1, typu 2, typu 3 i typu 17 (Th1, Th2, Th3, Th17), które promują i kontrolują odpowiedzi komórkowe i przeciwciała. Obserwowany jest wzrost wyjściowych poziomów neopteryny w osoczu, rozpuszczalnego markera aktywacji limfocytów T przez interferon-gamma.
Infekcja indukuje autoreaktywne limfocyty T i B (w tym komórki plazmatyczne wytwarzające przeciwciała). Te limfocyty B (lub ich przeciwciała i cytokiny) przekraczają barierę krew-mózg i zmieniają neurotransmisję, powodując charakterystyczny fenotyp.
Większość patogennych autoprzeciwciał w zaburzeniach neurologicznych wiąże się z białkami lub receptorami na powierzchni komórek neuronów, aksonów lub śródbłonka (czy też mieliście przeciwciała anty-neuroendothelium?). Konformacja tych autoantygenów jest ważna w wiązaniu autoprzeciwciała z autoantygenami. Dlatego metody wykrywania autoprzeciwciał, które zakłócają konformację białek podczas przygotowania, mogą prowadzić do mylących wyników. Do chwili obecnej większość metod immunologicznych wykorzystywała metody zakłócające strukturę białek. Na przykład metoda Western blot obejmuje homogenizację i detergenty, które uwalniają antygeny cytoplazmatyczne, rozplatają białka i rozrywają wiązania dwusiarczkowe. Z kolei w ostatnich odkryciach patogennych autoprzeciwciał wykorzystano systemy żywych komórek, które eksprymują potencjalne autoantygeny w ich naturalnym stanie konformacyjnym na powierzchni komórki.
W badaniach immunofluorescencji pośredniej (IF) wykorzystano cienkie skrawki tkanki mózgowej. Surowicę pacjenta inkubuje się na tkance, przemywa i wykrywa wiązanie przeciwciał pacjenta przy użyciu drugorzędowego przeciwciała antyludzkiego ze znacznikiem fluorescencyjnym i mikroskopii. Metodą tą można wykazać obecność autoprzeciwciał oraz regionalną i komórkową lokalizację autoantygenów. Pacjenci mają autoprzeciwciała przeciwko białkom cytoplazmatycznym neuronów (szczególnie tych w zwojach podstawy mózgu).
Idąc tym tropem, w ostatnich latach opublikowano wstępne prace nad domniemanymi autoantygenami, na które oddziałują te przeciwciała przeciwneuronalne. Autoantygeny o masach 40, 45 i 60 kDa zidentyfikowane metodą Churcha zidentyfikowano następnie jako enzymy glikolityczne (aldolaza C, enolaza specyficzna dla neuronów, enolaza nieneuronalna i kinaza pirogronianowa M1). Te enzymy glikolityczne są obecne w cytoplazmie neuronów i na powierzchni komórek neuronów. Biorą udział w metabolizmie energetycznym i wspierają kanały jonowe. Co więcej, te same enzymy glikolityczne występują na powierzchni komórek bakterii paciorkowcowych. Nieneuronalną enolazę uznano już wcześniej za autoantygen w gorączce reumatycznej. Niezależna grupa badaczy zidentyfikowała następnie kinazę pirogronianową M1 jako autoantygen w zespole Tourette, która stwierdziła podwyższone stężenie przeciwciał wobec kinazy przeciwpirogronianowej podczas zaostrzeń choroby wywołanych przez paciorkowce.
Kirvan i in. stwierdzili reaktywność krzyżową autoprzeciwciał pomiędzy paciorkowcową N-acetylo-glukozaminą i lizogangliozydem mózgowym; przeciwciała przeciwko lizogangliozydom były podwyższone w pląsawicy i PANDAS. Co więcej, wydaje się, że te autoprzeciwciała powodują zmiany w aktywacji kinazy wapniowo-kalmodulinowej II i prawdopodobnie zwiększenie poziomu hydroksylazy tyrozynowej (a tym samym syntezy dopaminy), co teoretycznie może skutkować pojawieniem się tików.
Obecność autoprzeciwciał nie jest nadal wystarczająca do udowodnienia związku przyczynowego. Na przykład przeciwciała przeciwmózgowe wykryto w chorobie Huntingtona i genetycznej chorobie Parkinsona, gdzie mogą być wytwarzane w odpowiedzi na chorobę, a nie być przyczyną. Aby autoprzeciwciała można było uznać za chorobotwórcze, powinny być również obecne w narządzie docelowym, bierny transfer autoprzeciwciał powinien wywołać chorobę u zwierząt, a objawy pacjentów powinny złagodnieć po usunięciu autoprzeciwciał.
Uszkodzenia za pośrednictwem przeciwciał mogą mieć charakter zapalny; alternatywnie autoprzeciwciała mogą działać jako „neurotoksyny”, powodując upośledzenie wewnątrzkomórkowej sygnalizacji neuronalnej. Zgodnie z hipotezą, że autoimmunologiczna odpowiedź zapalna w zwojach podstawy mózgu może wystąpić w podgrupie pacjentów z tikami lub objawami zespołu obsesyjno-kompulsywnego (ZOK), badania z wykorzystaniem rezonansu magnetycznego wykazały powiększenie zwojów podstawy mózgu zarówno w przypadkach pląsawicy, jak i PANDAS. Podobne donoszono o nagłym wystąpieniu zaburzeń obsesyjno-kompulsyjnych związanych z nowo nabytą infekcją Mycoplasma pneumoniae. Odkrycia te mogą mieć związek z ostrymi zmianami zapalnymi występującymi wkrótce po ich wystąpieniu, ponieważ wolumetria nie różniła się pod względem obecności autoprzeciwciał u pacjentów z dłuższym czas trwania choroby. Zgodnie z hipotezą „neurotoksyczności”, Teixeira i in. odkryli zwiększony napływ wapnia do linii komórkowych nerwiaka po inkubacji przeciwciał od pacjentów.
Oprócz mechanizmów, w których pośredniczą przeciwciała, objawy obserwowane u pacjentów z PANDAS, takie jak tiki, objawy ZOK i objawy depresyjne/lękowe, mogą być również bezpośrednio lub pośrednio wywoływane przez cytokiny. Patologicznym modelem tych procesów są zachowania chorobowe i „depresja wywołana cytokinami”. Cytokiny prozapalne, w tym TNF-α, IL-1β i IL-6, IL-17a (mozna je zbadac w Alab), mogą modulować transmisję katecholaminergiczną, zmieniać metabolizm tryptofanu, prowadząc w ten sposób do nieprawidłowego metabolizmu serotoniny i nadmiernej produkcji potencjalnie toksycznych metabolitów tryptofanu (o co często proszę, aby sprawdzać w panelu NEURO z Alab), zmieniać ekspresję genów, i aktywować oś podwzgórze-przysadka-nadnercza, co prowadzi do nieprawidłowej reakcji na stres.
Istnieją przekonujące dowody na występowanie stanu hiperdopaminergicznego ze zwiększoną aktywnością dopaminergiczną w prążkowiu i zwiększone wewnątrzsynaptyczne uwalnianie dopaminy w prążkowiu o stymulacji.
Rozwijając koncepcję pierwotnie wyrażoną przez Cunninghama, Kawikową, Bothwella i Leckmana (Leckman JF i wsp., komunikacja osobista) zaproponowano model patofizjologiczny wyjaśniający udział mechanizmów immunologicznych w patofizjologii tików i zaburzeń pokrewnych. Zwiększone narażenie pacjentów na zakażenia może indukować syntezę krzyżowo reagujących przeciwciał przeciwnerwowych, np. przeciwciała antylizogangliozydowe, które promują syntezę dopaminy i egzocytozę poprzez aktywację kinazy CaM II. Przeciwciała przeciwko enzymom antyglikolitycznym mogą również modyfikować pobudliwość neuronów i wpływać na uwalnianie neuroprzekaźników, chociaż obecnie brakuje dowodów na poparcie tej hipotezy. Jednocześnie zwiększone narażenie na zakażenia może sprzyjać odpowiedziom zapalnym pierwszego rzutu, które mogą nasilać uwalnianie dopaminy przez włókna autonomiczne i wzmacniać modulację dopaminergiczną obwodowych komórek odpornościowych. Dopamina wywiera działanie immunomodulujące poprzez aktywację receptorów dopaminy D1/D5 na powierzchni komórek limfocytów, powodując funkcjonalną regulację w dół limfocytów Treg, aktywację spoczynkowych limfocytów T i zwiększone uwalnianie cytokin (szczególnie TNF-α). Treg regulacja w dół wypaczyłaby mechanizmy regulacji odporności na korzyść autoimmunizacji, podczas gdy wzrost TNFα zwiększyłby przepuszczalność bariery krew-mózg. Zmiany te mogą prowadzić do wzmożonej odpowiedzi autoimmunologicznej i jeszcze większego uwalniania dopaminy w zwojach podstawy mózgu, co z kolei przyczynia się do wystąpienia klinicznych objawów.
Rysunek 1 podsumowuje proponowany mechanizm (jest on propozycją przedstawioną w artykule o Zespole Tourette, jak dla mnie niezdiagnozowanym PANDAS w większości przypadków).
Odkryto, że receptory Toll-podobne (TLR) są kluczowymi receptorami rozpoznawania wzorców (PRR) zaangażowanymi w rozpoznawanie wzorców molekularnych związanych z patogenami (PAMP). Późniejsze badania wykazały ich udział w rozpoznawaniu różnych wzorców molekularnych związanych z uszkodzeniami/zagrożeniami (DAMP) generowanych przez samego gospodarza. Zatem receptory TLR są zdolne do rozpoznawania szerokiego zakresu wzorców/cząsteczek pochodzących od patogenów i gospodarza, jak również inicjowania prozapalnej odpowiedzi immunologicznej poprzez aktywację NF-κB i innych czynników transkrypcyjnych powodujących syntezę cząsteczek prozapalnych. Proces zapalenia nerwów występuje zarówno w jałowych, jak i zakaźnych chorobach zapalnych ośrodkowego układu nerwowego (OUN) i może prowadzić do rozwoju neurodegeneracji.
Uszkodzenie mózgu prowadzi do aktywacji szlaków naprawczych, takich jak odbudowa sieci neuronowej, zwiększenie plastyczności i zapewnienie alternatywnych metod wykonywania zadań dotkniętych utratą tkanki mózgowej. Zapalenie układu nerwowego to reakcja komórek glejowych (np. mikrogleju i astrocytów) na infekcję lub uraz w ramach wrodzonego układu odpornościowego. Aktywowane komórki glejowe wytwarzają prozapalne cytokiny, enzymy i cząsteczki adhezyjne – jest to odpowiedź wspomagana przez receptory TLR.
Ścieżka TLR - teraz będzie coś dla biologów i pasjonatów :)
Po rozpoznaniu PAMP, sygnalizacja TLR odbywa się albo w sposób zależny od MyD88, albo niezależny od MyD88/zależny od Trif. MyD88 jest białkiem adaptorowym, które oddziałuje głównie z białkami TIRAP/MyD88-adapteropodobnymi (Mal) i TRAM i odgrywa kluczową rolę w umożliwieniu rekrutacji kinaz związanych z IL-1R (IRAK)-1, 2, 3, 4. TIRAP/Mal i TRAM są również wymagane do aktywacji sygnalizacji zależnej od TRIF. Wszystkie receptory TLR sygnalizują szlak zależny od MyD88, z wyjątkiem TLR3, który sygnalizuje wyłącznie poprzez TRIF.
Kluczowe etapy sygnalizacji TLR za pośrednictwem MyD88 i TRIF zostały opisane poniżej na przykładzie TLR4. Wynikiem obu szlaków jest aktywacja IRF3, NF-kB i AB-1, które regulują ekspresję cytokin prozapalnych i IFN typu 1.
Szlak zależny od MyD88:
- TLR4 jest stymulowany przez ligand.
- MyD88 jest rekrutowany za pośrednictwem TIRAP.
- MyD88 rekrutuje kinazy IRAK (głównie IRAK4).
- IRAK są fosforylowane, a następnie aktywowane.
- IRAKi odłączają się od MyD88.
- IRAK oddziałują z ligazą E3 TRAF6.
- TRAF6 tworzy kompleks z enzymami koniugującymi ubikwitynę UBE2N/UBC13 i UBE2V1/UEV1A.
- Ten kompleks poliubikwitynuje białka docelowe, takie jak TAK1/TAB1/TAB2/TAB3.
- Poliubikwitynacja prowadzi do aktywacji kompleksu TAK, a następnie aktywacji IKK.
- IkappaB są niszczone przez proteasom 26S, co powoduje translokację NF-kB do jądra i regulację genów zależnych od NF-kB.
- Sygnalizacja kinazy MAP jest również aktywowana przez kompleks TAK, co powoduje indukcję cytokin prozapalnych poprzez aktywację AP-1.
Szlak niezależny od MyD88/zależny od TRIF:
Ścieżka 1
- TLR4 jest stymulowany przez ligand.
- TLR4 rekrutuje TRAM i TRIF.
- TRIF współdziała z TBK1.
- Kinaza TBK1 i IKKi umożliwiają fosforylację IRF3.
- Fosforylacja IRF3 powoduje jego aktywację i translokację do jądra, gdzie pełni rolę czynnika transkrypcyjnego.
Ścieżka 2
- TLR4 jest stymulowany przez ligand.
- TRIF współdziała z TRAF6 i RIP1.
- Powoduje to zniszczenie IkappaB przez proteasom 26S i późniejszą aktywację NF-kB.
TLR i komórki mózgowe
Mikroglej to komórki podobne do makrofagów obecne w ośrodkowym układzie nerwowym (OUN), które pośredniczą w neuronalnych interakcjach immunologicznych. Wydaje się, że aktywowany mikroglej pełni głównie rolę neuroprotekcyjną w przypadku ostrego uszkodzenia. Warunki stresowe, takie jak niedotlenienie, powodują zwiększoną sygnalizację TLR w mikrogleju. Oprócz TLR10, wszystkie TLR ulegają ekspresji w mikrogleju (Tabela 1). Na przykład TLR4 i TLR2 powiązano z uwrażliwianiem na apoptozę mikrogleju, co sugeruje rolę w zapobieganiu nadmiernemu zapaleniu. Mikroglej wytwarza prozapalne cytokiny, enzymy i cząsteczki adhezyjne, które inicjują migrację leukocytów przez barierę krew-mózg i promują funkcje efektorowe w tych naciekających komórkach.
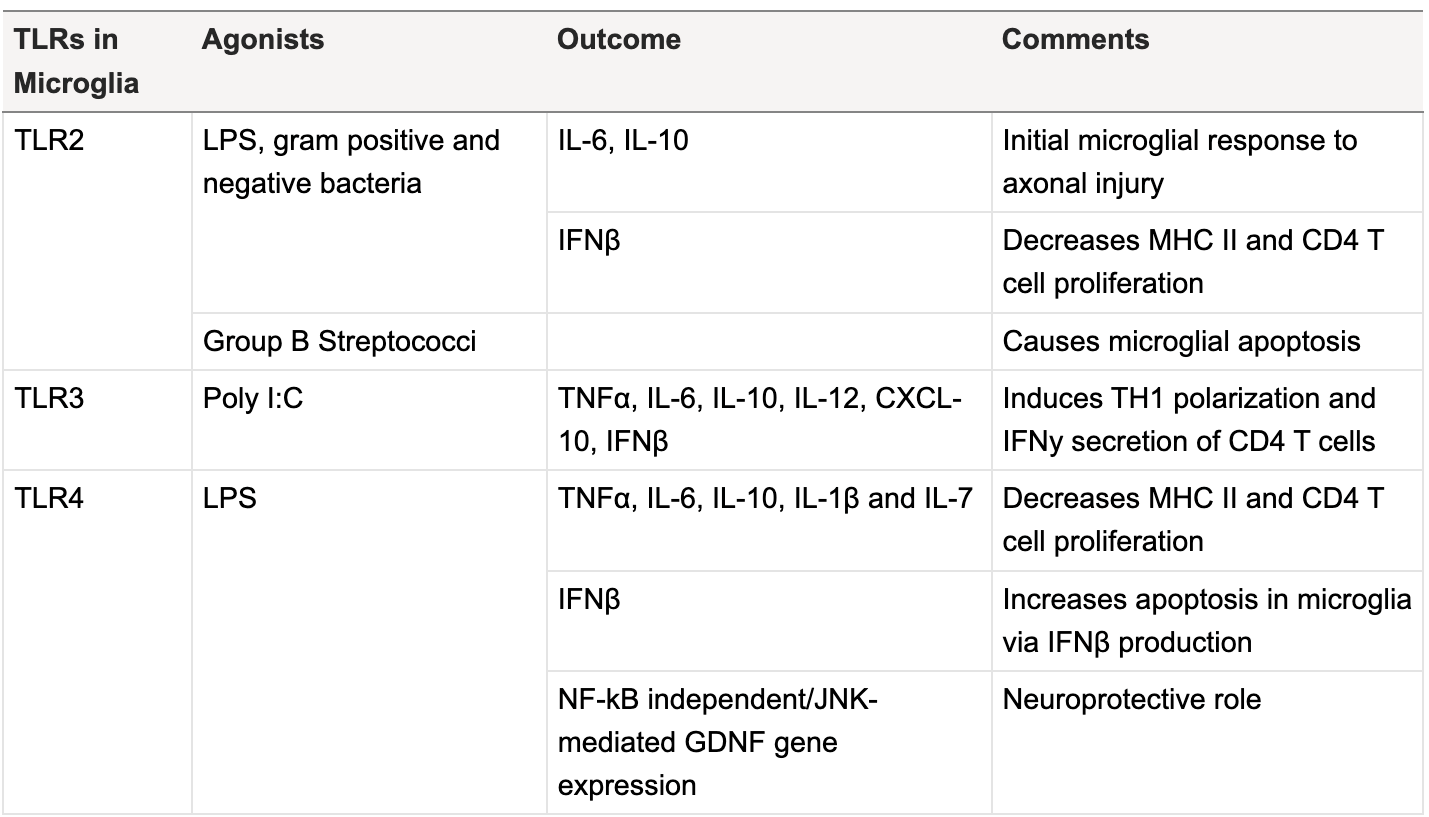
Ludzkie astrocyty wyrażają TLR1-7, 9 i 10 (patrz tabela 2). Leki takie jak statyny zwiększają wytwarzanie cytokin przez astrocyty za pośrednictwem TLR4 i są powiązane z neuropatiami. Z kolei cytokiny i agoniści TLR zwiększają także ekspresję ligandów chemokin, takich jak CCL2, CCL3 i CCL5, które następnie przyciągają komórki odpornościowe do miejsca zapalenia.
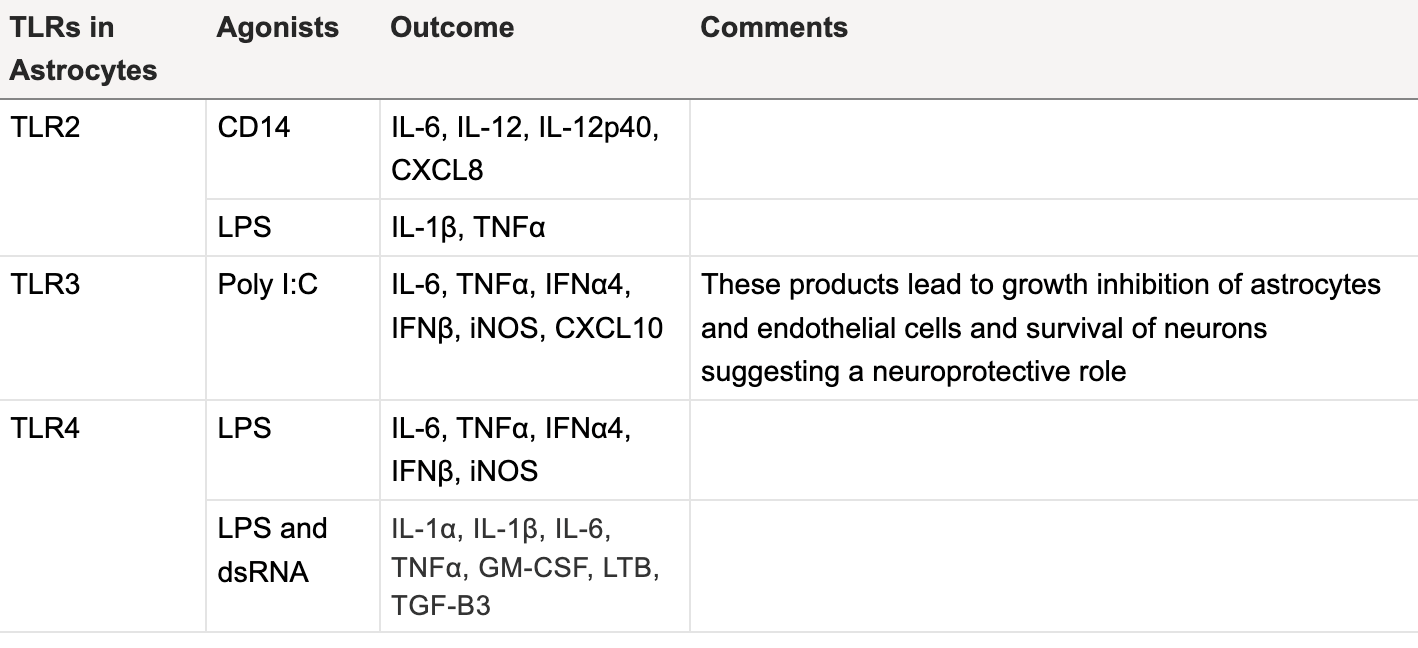
Niewłaściwą aktywację receptorów TLR, takich jak TLR4, i dalszych szlaków powiązano z niektórymi chorobami autoimmunologicznymi, w tym stwardnieniem rozsianym.
Istnieje coraz więcej dowodów na znaczenie nieprawidłowej ekspresji neuronów TLR w rozwoju stanów patologicznych. Wyraźną różnicę widać w aktywacji TLR w zróżnicowanych neuronach w porównaniu z neuronalnymi komórkami progenitorowymi, chociaż oba typy komórek wyrażają TLR2 i TLR4. TLR2 bierze udział w neurogenezie hipokampa, podczas gdy TLR4 zmniejsza proliferację i różnicowanie neuronów. Obydwa receptory TLR regulują los komórek poprzez szlaki MyD88 i NF-kB, jednakże jest to specyficzne dla komórek progenitorowych, mediatory zróżnicowanych komórek są nieznane.
Bakterie stymulują odpowiedź Th17 przez receptory TLR, NOD-podobne (NLR) i lektynowe typu C (CLR). Aktywowane limfocyty Th17 wytwarzają liczne cytokiny, głównie interleukinę 17 oraz chemokiny, które przyciągają i pobudzają fagocyty celem eliminacji bakterii. W ten sposób komórki Th17 przyczyniają się do rozwoju odporności ochronnej, zwłaszcza przeciwko bakteriom zewnątrzkomórkowym: Staphylococcus aureus, Streptococcus pneumoniae i Klebsiella pneumoniae.
S. pyogenes, należący do grupy A paciorkowców (GAS), jest czynnikiem etiologicznym m.in. szkarlatyny, anginy, paciorkowcowego zespołu wstrząsu toksycznego oraz zapalenia ucha środkowego u ludzi. GAS podane myszom donosowo indukują różnicowanie limfocytów T w kierunku Th17 w tkance limfatycznej związanej z nosem i gardłem, co przyczynia się do stymulacji ochronnej odpowiedzi immunologicznej. Przewlekłe zakażenie paciorkowcem prowadziło bowiem do wytwarzania IL-6 i TGF-β1 w NALT, a cytokiny te są niezbędne w przebiegu różnicowania limfocytów T w kierunku Th17.
Dileepan i wsp. wykazali, że indukcja odpowiedzi immunologicznej z udziałem limfocytów Th17 u myszy zakażonych GAS uzależniona jest od drogi zakażenia oraz obecności IL-6, natomiast nie zależy od superantygenu paciorkowcowego (epitop 2W białka M). Następstwem zakażeń S. pyogenes u dzieci jest często reumatoidalne zapalenie stawów i/lub zapalenie mięśnia sercowego. Joosten i wsp. opracowali model myszy z miejscowym, przewlekłym, uszkadzającym zapaleniem stawów, wywołanym kilkukrotnym śródstawowym podaniem składowych ściany komórkowej S. pyogenes. Wykorzystując ten model doświadczalny u zwierząt pozbawionych genów dla cytokin wskazano na główną rolę IL-17 i IL-1β w rozwoju przewlekłej fazy choroby, podczas gdy TNF-α nie miał wpływu na jej przebieg. Co więcej, u myszy IL- -17R–/–, RAG-2–/– i IFN-γ–/– nie rozwijało się przewlekłe zapalenie stawów. Te badania wyraźnie wskazują na istotną rolę IL-17 i IL-1β w rozwoju przewlekłego zapalenia stawów u myszy w odpowiedzi na składniki ściany komórkowej S. pyogenes. Nie jest wykluczone, że obie cytokiny mają podobne znaczenie w rozwoju reumatoidalnego zapalenia stawów u ludzi, powstałego w wyniku paciorkowcowych angin.
W późniejszych badaniach odkryto, że komórki Th17 i wytwarzana przez nie IL-17, hamują kolonizację błony śluzowej nosogardzieli przez pneumokoki oraz ograniczają rozwój paciorkowcowego zakażenia u myszy. Ochrona przed kolonizacją błony śluzowej jest bardzo zależna od aktywności neutrofilów, natomiast przy braku receptora dla IL-17 jest zniesiona. Co więcej, rekombinowana ludzka IL-17 zwiększa zabijanie komórek S. pneumoniae przez ludzkie neutrofile krwi obwodowej w warunkach in vitro niezależnie od przeciwciał przeciwotoczkowych. Wykazano też, że antygeny pneumokoków mogą pobudzić wytwarzanie IL-17 przez komórki migdałków u dzieci oraz przez komórki jednojądrzaste krwi obwodowej zdrowych dorosłych ochotników, nie stymulują natomiast komórek krwi pępowinowej.
Aktywacja receptorów TLR powoduje produkcję cytokin, w tym il-17, chemokin i interferonów wraz z aktywacją czynnika transkrypcyjnego NF-κB, co skutkuje zaburzeniami zapalnymi. Szlak sygnalizacyjny TLR4 jest najdokładniej zbadanym szlakiem TLR. Zatem ukierunkowanie na modulację TLR4 może stanowić podejście terapeutyczne w celu złagodzenia chorób zapalnych, które są podkreślone przez kaskadę zapalną. Rośliny lecznicze o działaniu przeciwzapalnym wykazują cenne działanie i są stosowane jako alternatywne źródła łagodzenia chorób związanych ze stanami zapalnymi. Flawonoidy i inne fitochemikalia mają obiecujące działanie farmakologiczne ze względu na ich stosunkowo niski koszt i wysoki profil bezpieczeństwa.
Wiadomo, że apigenina, naturalny flawonoid występujący w wielu roślinach, owocach, warzywach, ziołach i przyprawach, ma właściwości przeciwutleniające i przeciwzapalne, co jest widoczne w stosowaniu tych substancji od wieków w celach leczniczych w leczeniu astmy, bezsenności, chorobie Parkinsona, nerwobólach i półpascu. Jednakże istnieje znaczny niedostatek informacji na temat wpływu na komórki odpornościowe, zwłaszcza komórki dendrytyczne (DC), które utrzymują krytyczną równowagę pomiędzy immunogenną i tolerogenną odpowiedzią immunologiczną, w immunospecjalizowanym miejscu, takim jak centralny układ nerwowy (OUN). In vivo, w mysich modelach stwardnienia rozsianego C57BL/6 (postępującego) i SJL/J (remisyjnego) stwardnienia rozsianego po leczeniu Apigeniną zaobserwowano znaczące zmniejszenie nasilenia eksperymentalnego autoimmunologicznego zapalenia mózgu i rdzenia (EAE), progresji i nawrotu. Myszy EAE leczone apigeniną wykazywały zmniejszoną ekspresję integryny α4 i CLEC12A na DC śledziony i zwiększoną retencję komórek odpornościowych na obwodzie w porównaniu z nieleczonymi myszami EAE. Korelowało to w konsekwencji z wynikami immunohistochemii dotyczącymi zmniejszonego nacieku komórek odpornościowych i zmniejszonej demielinizacji w OUN. Wyniki te wskazują na ochronną rolę Apigeniny przeciwko efektom neurodegeneracyjnym wynikającym z wejścia stymulowanych DC patogennych komórek T do OUN, co sugeruje potencjalną terapię dla choroby neurozapalnej. Apigenina ma działanie przeciwzapalne, może zmniejszać wytwarzanie mediatorów stanu zapalnego, które jest regulowane przez zależną od receptora Toll-podobnego 4 aktywację szlaków Akt, mTOR i NF-κB oraz aktywację JNK i p38-MAPK. Apigenina hamuje produkcję cytokin IL-1β i IL-6 oraz chemokin CCL17 i CCL27; ekspresje cyklooksygenazy-2; wzrost poziomu receptora Toll-podobnego-4, fosforylowanego Akt i mTOR; aktywacje NF-κB; aktywacje JNK i p38-MAPK; oraz wytwarzanie reaktywnych form tlenu/azotu.
Badania funkcjonalne in vitro wykazały, że apigenina zmniejsza indukowaną przez LPS i IFN-γ produkcję czynnika zapalnego mikrogleju oraz aktywację M1 poprzez szlak TLR4/MyD88.
Apigenina hamuje prezentację autoantygenu i funkcje stymulujące APC niezbędne do aktywacji i ekspansji autoreaktywnych komórek Th1 i Th17 oraz komórek B w toczniu. Apigenina powoduje także apoptozę nadaktywnych APC oraz komórek T i B tocznia, prawdopodobnie poprzez hamowanie ekspresji cząsteczek antyapoptotycznych regulowanych przez NF-κB, zwłaszcza COX-2 i c-FLIP, które ulegają trwałej nadmiernej ekspresji w komórkach odpornościowych tocznia. Zwiększanie biodostępności roślinnych inhibitorów COX-2 i NF-κB, takich jak apigenina, może być cenne w hamowaniu stanu zapalnego w toczniu i innych chorobach, w których pośredniczy Th17, takich jak reumatoidalne zapalenie stawów, choroba Leśniowskiego-Crohna i łuszczyca, a także w zapobieganiu stanom zapalnym w układzie nerwowym.
Co najważniejsze apigenina wzmaga prawidłowe działanie receptorów dopaminowych - zadziała u osób mających przeciwciała na tych receptorach.
Na uwagę zasługuje fakt, że należy używać jej ostrożnie u osób z zespołem Wilsona.
Dawki terapeutyczne zaczynają się od 10 mg/kg mc czystej apigeniny do 50 mg/kg mc w chorobach neurologicznych.
Żywność i napoje zawierające Apigeninę:
- pietruszka
- kolendra
- szpinak
- seler
- cebule
- pomarańcze
- grejpfrut (suplement diety https://protonlabs.pl/products/apigenina-czysty-proszek-pure-apigenin, https://www.mcsformulas.com/vitamins-supplements/apigenin-pro-liposomal/?gclid=CjwKCAjw3dCnBhBCEiwAVvLcu5CRz4JHpYoCIWPjcELdzRgx0p7xe7htFa7kPDP40nIbwigCH-MhoBoCi-cQAvD_BwE)
- kumkwaty
- brukiew
- rumianek (suplement diety https://eternalis.pl/pl/strona-glowna/36-48-apigenina-proszek-5905090423676.html#/1-wielkosc_opakowania-15_g, https://ekstrakty.com/relaks-i-sen/rumianek-pospolity-ekstrakt-98-apigeniny-10g)
- tymianek
- oregano
- estragon
- kwiat pasji
- mięta
- bazylia
- herbata
Zwiększone uwalnianie i ekspresja cytokin zapalnych, w tym interleukiny-1β, czynnika martwicy nowotworu-α, przez LPS, zostały znacznie zmniejszone przez genisteinę. Genisteina osłabiała również indukowaną przez LPS generację reaktywnych form tlenu i za pośrednictwem LPS translokację jądrową jądrowego czynnika kappa B (NF-κB), związaną z blokowaniem degradacji inhibitora NF-κB-α. Ponadto genisteina silnie hamowała wiązanie LPS z powierzchnią komórki mikrogleju, co wskazuje na antagonistyczne działanie genisteiny przeciwko receptorowi Toll-like 4 (TLR4) i hamowała indukowaną przez LPS ekspresję TLR4.
Zanim więc parskniesz śmiechem na swojej relacji w mediach społecznościowych, że uczę rodziców jak pomóc dziecku na podstawie badań na zwierzętach, zastanów się na kim testuje się leki zanim trafiają do naszych brzuchów?